Current projects at XTBG
Since I arrived at XTBG in June 2012 as PI of the Ecological Evolution group,
I have been focusing on the mechanisms of diversification of tropical taxa, and
on the role of adaptation and hybridization in speciation using Ficus and
Fagaceae as model species.
I also develop bioinformatic tools for the analysis of Next-Generation Sequencing data.
(see here for available tools).
I am supervising several postdoc and student projects:
- Development of molecular markers and phylogeny of Ficus: Postdoc project of Marie Fougère-Danezan
- Development of molecular markers and phylogeny of Fagaceae: Postdoc project of Joeri S. Strijk
- Assembly and Alignment Free methods for phylogenetic reconstruction: PhD project of Fan Huan in collaboration with Chuck Cannon (TTU), and Anthony R. Ives (UW-Madison)
- Transcriptome analysis of cynipid wasp induced galls: PhD project of Warin Harrison in collaboration with Chuck Cannon (TTU)
- Using metabarcoding techniques for community analysis of litter arthropods: Master project of Shen Xianhui
For more information on the Ecological Evolution group, see here
Research direction 1: The role of natural selection and local adaptation in speciation
Speciation and reinforcement in Neotropical spiral gingers.
I also work on Neotropical spiral gingers (genus Costus), a group of plants that went through one of the fastest radiations known after the colonization of the Neotropics. In particular, I investigated the two species C. pulverulentus and C. scaber that diverged recently and share a large area of sympatry. These species present isolation mechanisms compatible with a model of speciation with reinforcement, also suggesting a role of natural selection. The objective of this project is to test the reinforcement hypothesis using two approaches:
- Understand the historical relationships between these two species, their genetic structure, and the patterns of current and historical gene flow, using microsatellite and RADseq markers
- Understand the genetic mechanisms of reproductive isolation by building a genetic map of these species, and mapping QTL responsible for isolation traits.
This work led by K.M. Kay at UCSC is still ongoing, but a genetic map containing 1275 RAD markers distributed over nine linkage groups (the number of chromosomes in Costus) has been built and the QTL mapping is under way.
Speciation, phylogeography and adaptive radiation of Caribbean lizards
 |
 |
 |
 |
During my postdoctoral position at Bangor University with Prof Roger S. Thorpe,
I have been investigating
the relative role of geographic isolation (allopatry) and natural selection in
population differentiation and speciation. The island of Martinique (in the
French West Indies) and its endemic anole, Anolis roquet offer a great
opportunity to disentangle the relative role of these two factors. Indeed,
Martinique once was composed of five precursor islands that joined to form
present day Martinique about two million years ago. Four distinct lineages
of A. roquet evolved in allopatry on these precursor islands for up
to 8 million years before coming into secondary contact when the islands joined.
Furthermore Martinique is a mountainous island and is characterised by very
sharp ecological gradients from dry coastal scrubland to wet mountain rainforest.
Anoles living in these different habitats are subject to very different selective
pressures and differ greatly morphologically. This system allows us to measure
population differentiation across lineage and habitat contact zones and hence
to look independently at the effect of these two factors.
Using mitochondrial DNA, microsatellites and morphology, we measured the
population differentiation along independent transects across habitat and/or
lineage contact zones. In all but one transects, we demonstrated that the
geological history had no effect on the population differentiation, while
habitat differences led to a strong reduction in gene flow. We concluded that
contrarily to the expectations, this lizard had a better fit to the ecological
speciation than to the allopatric speciation model, pointing to an important
role of natural selection in speciation.
To investigate the generality of the patterns observed in Martinique, I am using the other species of the roquet group (A. luciae in St Lucia, A. griseus and A. trinitatis in St Vincent, A. aeneus and A. richardii in Grenada and A. bonairensis in Bonaire) as natural replicates. I am also interested in the phylogenetic relationships of the roquet group and for islands with two species of anoles (St Vincent and Grenada) to the comparative phylogeography of these species.
Finally I am also interested in phylogeny and adaptive radiation of the dwarf geckos Sphaerodactylus fantasticus and Sphaerodactylus vincenti in the Lesser Antilles.
Research direction 2: Tool development in evolutionary genetics of non-model organisms.
Evolutionary genetics work on non-model organisms has often been limited by the lack of genomic resources or of bioinformatic tools adapted to the analysis of species for which no reference genome exist.
For instance, the analysis of gene expression by RNA sequencing requires as
a first step a de-novo assembly of the transcriptome, while a simple mapping
of reads to a reference genome is sufficient for model organisms. De-novo
transcriptome assembly of non model organisms present different challenges
from genome assembly, in particular the non even coverage between different
transcripts, while an even coverage is expected by genome assembly software. While
working at the University of Geneva with Dr. Juam Montoya-Burgos, I developed
two bioinformatic tools to
optimize the assembly of de-novo transcriptome sequencing of non-model organisms
from Next-Generation Sequencing (NGS) data. The first, called multiple-k method,
relies on the fact that different k-mer values are needed to assemble transcripts
with different coverage. The second, called Scaffolding using Translation Mapping
(STM), makes use of a reference proteome to help assemble transcriptomes. Since
sequence divergence at the amino-acid level is much lower than at the nucleotide
level, it is possible to use the known proteome of distant model species to
improve the transcriptome assembly of species with no reference.
The paper describing the methods and the source code of the pipeline are
available here
Another problem generally encountered when working on non-model organisms is the lack of a sufficient number of variable markers for phylogenomics and population genomics. The development of NGS technology is changing this situation, and I am currently developing two alternative methods based on the Illumina MiSeq platform to generate a large amount of sequence information in two plant groups, the genus Ficus, and the Fagaceae. These methods will generate markers usable for both multilocus phylogenetic approaches and population genomics.
I also develop bioinformatic tools for the analysis of Next-Generation Sequencing
data. For instance, because most tools available for RAD-seq analysis (or similar
methods) don't allow the recovery of full haplotypes at each RAD locus, important
information that could be used for phylogenetic analyses is lost. I am working
on a tool to extract full haplotype information from NGS reads that generates
input files for IMa2, a software making full use of the haplotype information.
Finally, I am working on a method to reconstruct phylogenetic relationships
directly from raw NGS reads without preliminary assembly or alignment.
Research direction 3: Evolution of viviparity in Zootoca vivipara
My PhD work was part of a wider project aimed at understanding the evolution of reproductive modes in the common lizard Zootoca vivipara. Indeed, this lizard is a rare example of vertebrate species exhibiting a reproductive bimodality, i.e. where both oviparous and viviparous populations coexist within a single species (see figure below). This scarce situation offers a great opportunity to study this major evolutionary transition.
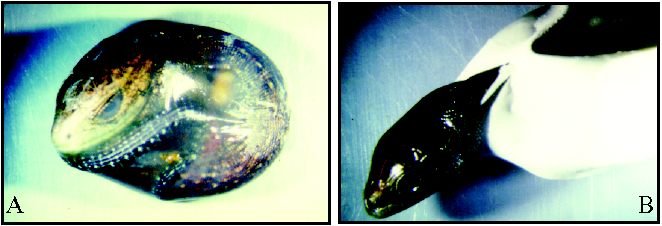
B) Hatching of the oviparous form, about 40 days after egg-laying.
Photos by Benoît Heulin.
The first part of the project consisted to understand the geographical
distribution of the various oviparous and viviparous populations of the European
common lizard (Zootoca vivipara), to identify the distinct lineages existing
in this species, and to elucidate the phylogenetic relationships of these
lineages. Using mitochondrial DNA, I identified two oviparous and four viviparous
lineages, and inferred that several shifts between reproductive modes occurred
in this species. Furthermore, the most parsimonious hypothesis suggests that
a reversal from viviparity back to oviparity occurred. However, more work
using nuclear markers is needed to confirm this hypothesis, since preliminary
analyses using AFLP suggested that introgression between the different lineages
might have obscured the evolutionary history of this species.
In parallel, we conducted functional investigation to understand the differences
between the oviparous and viviparous females. Both histological and
endocrinological studies showed that very little differences existed between
these two form, except for the eggshell characteristics, and the development
of oviduct glands (at the origin of the eggshell) during vitellogenesis.
To understand these differences, I am currently conducting a comparative
transcriptomic approach. The comparison of the transcriptional differences
between oviducts of oviparous and viviparous females should shed light on the
genetic mechanisms at the origin of the evolution of viviparity. This work is
conducted in collaboration with Dr Benoît Heulin (UMR CNRS 6553).